Table of contents
How to use bwa index
bwa index is the former step to use the command bwa mem. If you’d like to use multiple times of bwa meme, and every time you use bwa index at the same process. If the reference genome is big, it will take a long time. So we need to reuse the index like below
process bwa_index {
publishDir 'output'
container "staphb/bwa:latest"
input:
path ref from ref_ch7
output:
file 'chr1_GL383518v1_alt.fa.*' into bwa_index // we use `chr1_GL383518v1_alt.fa.*` to refer all the output files with the same prefix name
"""
bwa index $ref
"""
}
process reAlign_bwa {
publishDir 'output'
container "staphb/bwa:latest"
input:
file new_read1 from deplex_read1_ch
file new_read2 from deplex_read2_ch
path ref from ref_ch5
file '*' from bwa_index // we use '*' to refer all files in the Channel, thus this process folder will contain all the index files
// path path_ref from ref_path
output:
file 'realign.bam' into realign_ch
"""
bwa mem $ref $new_read1 $new_read2 > realign.bam
"""
}
The arrangement of process
Recently I always got the error that said there’s no such file that the command need to use. And I checked the output publishDir and found that the file existed in the folder. The process complains the error again and again. 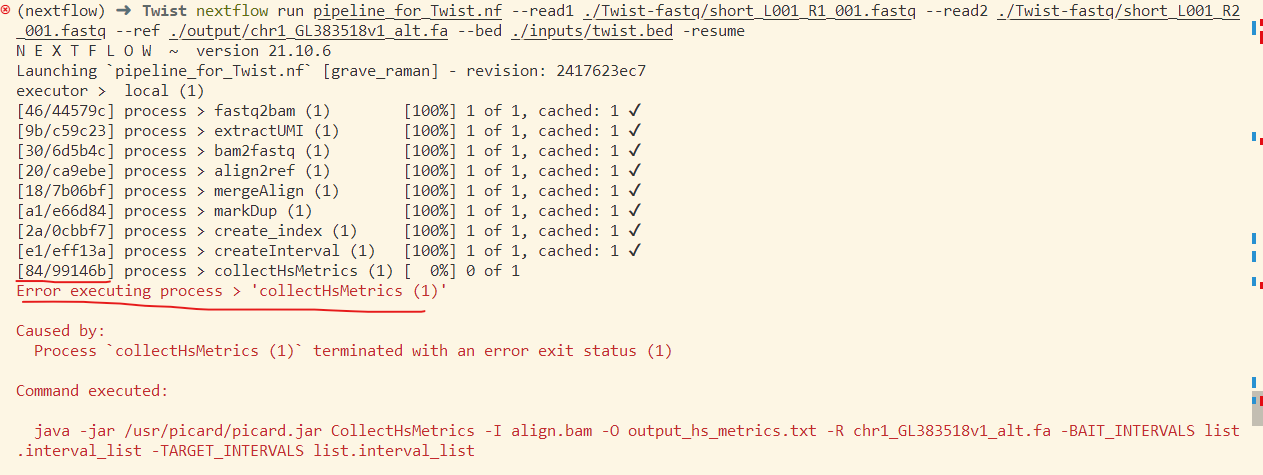
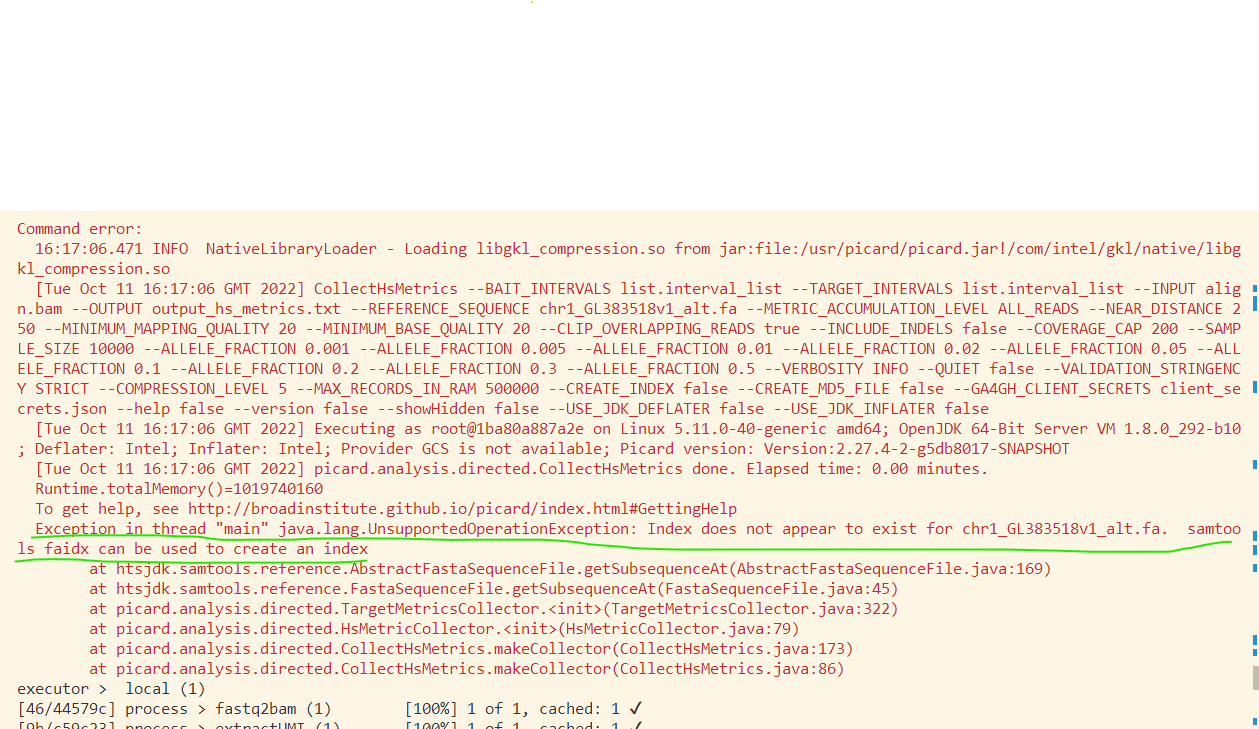
From the above picture, You can see that there’s noly one process failed. And it complains that some file didn’t exist. We should figure out where this setence means the file not exists. And at last I know that the place means the process folder, not the publishDir folder. See the first column of the executor. The first column is the position of this process like [2a/0cbbf7]. 2a is folder name in the work folder like below. 0cbbf7 is the subfolder. All the files this process needed are stored here. 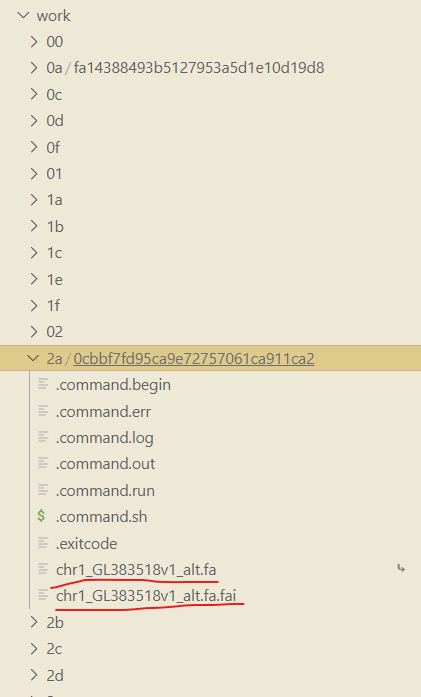
process create_index {
publishDir 'output'
container "staphb/samtools:latest"
input:
path ref from ref_ch3
output:
file 'chr1_GL383518v1_alt.fa.fai' into fai_ch
"""
samtools faidx $ref
"""
}
Now let’s see the failed process collectHsMetrics. It needs “chr1_GL383518v1_alt.fa.fai”. But it donesn’t exist in the process folder (align.bam chr1_GL383518v1_alt.fa list.interval_list). So we need to add it through the input like below
process collectHsMetrics {
publishDir 'output'
container 'broadinstitute/picard:latest'
input:
// file bed from bed_ch
file align_bam from align_ch2
path ref from ref_ch4
file interval from interval_ch
file index from fai_ch
output:
file "output_hs_metrics.txt"
"""
java -jar /usr/picard/picard.jar CollectHsMetrics -I $align_bam -O output_hs_metrics.txt -R $ref -BAIT_INTERVALS list.interval_list -TARGET_INTERVALS list.interval_list
"""
}
we added the index through the input file index from fai_ch. And you can generate the index in another process and just place it into a channel. So this process can get it throuh the channel.
Note: you should specify all the input files that you need to use in the command line. Because each process has a process folder, all the files that this process needed are saved in the process folder.
How to use the same file twice in two following process
Assign this file to different channels. And then you can use the file twice in the following channels.
process p1 {
output:
file 'hello_world.txt' into ch_a
file 'hello_world.txt' into ch_b
}
process p2 {
input:
file welcome from ch_a
}
process p3 {
input:
file welcome from ch_b
}
How to know the command that you should use in the docker container
- In the docker hub, search the image name and hit the tags. For example this address use picard.
- From the
IMAGE LAYERS, you may find a varialbe namedWORKDIR. In the picard, the WORKDIR isWORKDIR /usr/picard - In the terminal, try
sudo docker run broadinstitute/picard:latest java -jar /usr/picard/picard.jar FastqToSamto test the command - If the above method can not determine the command. Then you need to use the docker interface to try and find the command that you can use
sudo docker run -it container. Check folders like/usr/and/usr/binand/bin
How to use docker container in nextflow
- install
dockeron Ubuntu. Please refer this Website - create a new environment through
conda:conda create -n nextflow python=3.8 - activate the nextflow environment:
conda activate nextflow - install nextflow through conda :
conda install -c bioconda nextflow - create a
nextflow.configfile in the current project. About thenextflow.configfile please refer Official Website -
write docker setting in nextflow.config file. refer this link.If you’d like to use more different containers then you should write the nextflow.config like below.
fastq2bamandbam2vcfare the process names that you defined in the nextflow file. Within the{}, define the container that you want to use. And you also need to makedocker {enabled=true}. Please refer this website ```nextflow process { withName:fastq2bam { container = ‘python:3.8’ }withName:bam2vcf { container = ‘zlskidmore/fgbio:latest’ } }
docker { enabled = true } ```
- If you run the nextflow file now. You’ll get the error. Because you can’t run the docker directly. If you’d like to run docker in the terminal. Then you should use
sudo, likesudo docker run container. In this case, we don’t want to usesudoto run the docker. So we need to do the following stuff. Please refer this website - Create the docker group:
sudo groupadd docker - Add your user to the docker group :
sudo usermod -aG docker ${USER}. Here${USER}refer to the current user. Tryecho ${USER}to show the current user name - You would need to loog out and log back in so that your group membership is re-evaluated or type the following command:
su -s ${USER} - Verify that you can run docker commands without sudo.
docker run hello-world. If this command run successfully, then you can use the docker container in the nextflow now.
How to use python script in nextflow process
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
Channel.fromPath("/home/user/projects/nextflow/nf-training-public/nf-training/chr1_GL383518v1_alt.fa").into{file_ch ; bar; fool}
file_path = Channel.fromPath("/home/user/projects/nextflow/nf-training-public/nf-training/chr1_GL383518v1_alt.fa")
folder_path = projectDir
println folder_path
bar.subscribe {println it}
fool.view{"fool emit: " +it}
// fool.view()
process test_python {
publishDir 'out'
input:
path x from file_ch
val path_file from file_path
output:
stdout result
file 'output.txt'
"""
#!/home/user/miniconda3/envs/recon/bin/python
from pathlib import Path
print('$path_file')
print('$folder_path')
path_x = Path('$path_file')
print(path_x.parent)
with open('$x', 'r') as f:
list_line = f.readlines()
print(list_line[0])
with open('output.txt', 'w') as f:
f.write(list_line[0])
"""
}
result.subscribe {println it}
- if you want to assign the channel to multiple variables in nextflow, you can use
intooperator - If you just want to assign one variable just use equal sign
= projectDircan be used to as the UnixPath variable. It point to the folder where the nextflow script located in. And you can use it to specify other files in the same folder like'$folder_path/hello_world.txt'println folder_pathis to print the varilabe. If you’d like to print some value for confirmation, then you can useprintlndirectly- If you want to see the value of
Channel, then you’d have to usesubscribeandview. - If you want to use view to show the content of the Channel, then you use
(). If you’d like to use some pattern, then use{'some pattern :'+it} - define the nextflow process using keyword
processlikeprocess process_name { content } publishDir 'out': here nextflow will create a folder named “out” if there’s no such folder int theprojectDir. and all the output files that you define in theoutputwill be saved in the “out” folder.- define input variables through keyword
input. There are 7 qualifiers in input:val, env, file, path, stdin, tuple, each. More detailed information please check website.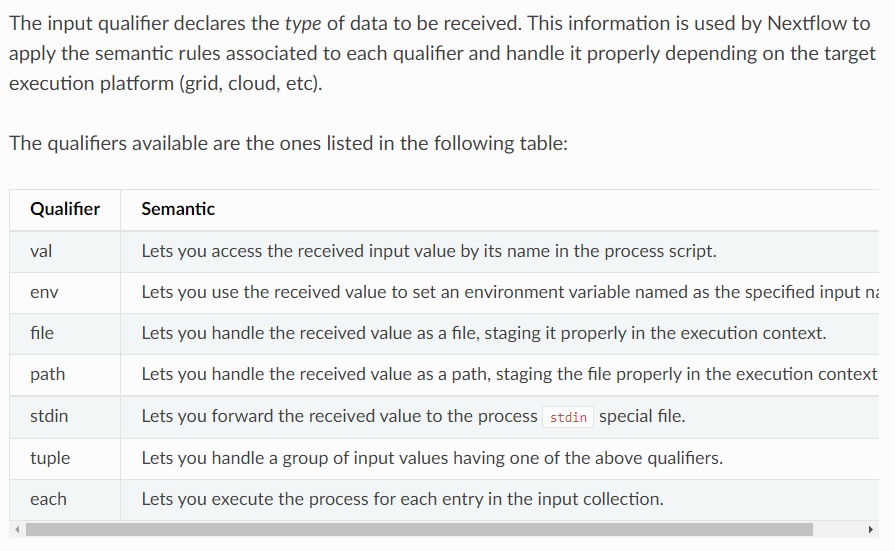
path x from file_ch: herefile_chis a channel. And the qualifier of the x ispath, thus it will contain all the UnixPath format information. Here if we replace the qualifier withfile, then it will only contain the file name. Acually it will only contain the stem of the filename like ‘Hell_world’ from ‘Hello_world.txt’.- define output variables through keyword
output. the qualifiers of output as shown below. Herefile 'output.txt'must be used in the python script. And the name must be kept same. Otherwise, nextflow will confuse and show errors.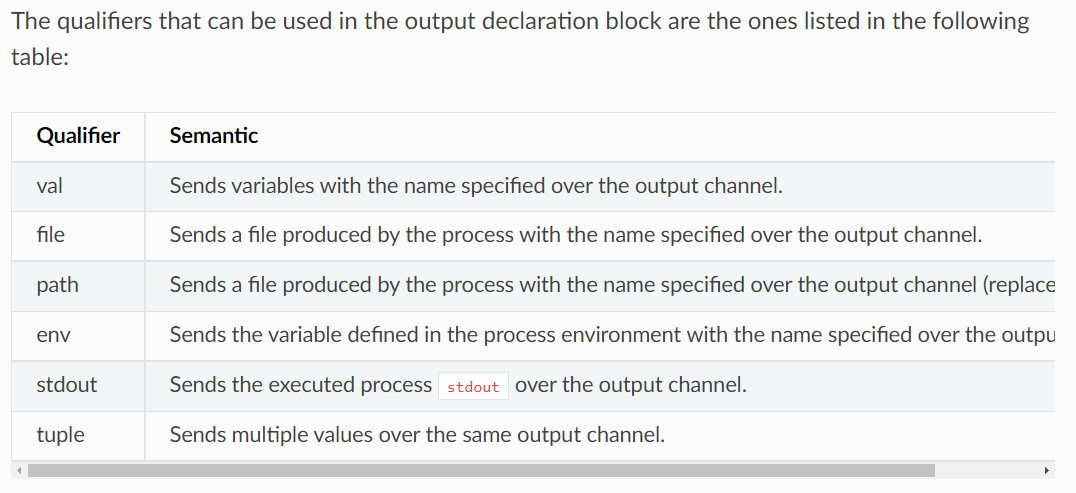
- If you’d like to show the results on the terminal, then you need to use
stdout. Actually instdout result, theresultacts as a channel that all the print stuff in the python script or other stuff show up in the terminal will be sent to the result as a queue. Thus we can usevieworsubscribeto show the content of the channel. - In the script, we need to specify the python environment through
#! - Actually the variables in the current process or the variables in the nextflow head part can be accessed through
'$variable'. Please remember that we use''or""to enclose the variable.